.svg)
Лікування гіпертрофічної кардіоміопатії (ГКМП)
.png)
Що таке гіпертрофічна кардіоміпатія
Гіпертрофічна кардіоміопатія (ГКМП) — це захворювання серцевого м'яза, при якому відбувається його потовщення (гіпертрофія) без очевидних причин, таких як гіпертензія чи вади клапанів. ГКМП має переважно генетичне походження і вражає близько 1 з 200–500 осіб, незалежно від віку та статі.
Хоча багато людей із ГКМП живуть повноцінно, захворювання може спричиняти серцеву недостатність, аритмії та є основною причиною раптової серцевої смерті у молодих людей і спортсменів.
При ГКМП стінка лівого шлуночка (рідше — правого) потовщується ≥15 мм у дорослих або ≥13 мм у родичів пацієнтів із підтвердженою ГКМП. У дітей діагностика ґрунтується на відхиленні товщини стінки від норми (Z-показник >2).
Найчастіше гіпертрофія вражає міжшлуночкову перегородку і має асиметричний характер. Однак можливе залучення інших ділянок серця.
.webp)






Типи гіпертрофічної кардіоміопатії: обструктивна та необструктивна форми
Гіпертрофічна кардіоміопатія (ГКМП) поділяється на два основних типи залежно від наявності або відсутності перешкоди кровотоку з лівого шлуночка:
Обструктивна форма (оГКМП)
Обструктивна ГКМП діагностується при наявності градієнта тиску у вихідному тракті лівого шлуночка (ВТЛШ) ≥30 мм рт. ст. у спокої або після провокаційних проб (наприклад, проба Вальсальви, фізичне навантаження).
- Градієнт ≥50 мм рт. ст. є показанням до розгляду інвазивного лікування.
- Причини: потовщена міжшлуночкова перегородка, систолічний передній рух (SAM) передньої стулки мітрального клапана.
- Зустрічається у близько 70% пацієнтів із симптомами.
- Типові симптоми: задишка при навантаженні, біль у грудях, запаморочення, непритомність.
Необструктивна форма (ноГКМП)
При необструктивній ГКМП значущої обструкції немає (градієнт тиску <30 мм рт. ст.).
- Причини симптомів: діастолічна дисфункція, мікросудинна ішемія, аритмії.
- Основні прояви: задишка, серцева недостатність зі збереженою фракцією викиду, порушення ритму.
Особливості діагностики
Обструкція може бути лабільною — виникати лише під час навантаження, після їжі чи при зміні положення тіла. Тому для точної діагностики потрібні провокаційні тести, зокрема ЕхоКГ з навантаженням.
Чому важливо визначити тип ГКМП?
Тип ГКМП визначає підхід до лікування:
- При обструктивній формі застосовуються: хірургічна міектомія, алкогольна абляція, інгібітори міозину (мавакамтен).
- При необструктивній формі основна мета — контроль симптомів серцевої недостатності та лікування аритмій.
Розподіл на обструктивну і необструктивну форми є першим кроком до побудови індивідуального плану ведення пацієнта.
Причини виникнення та фактори ризику гіпертрофічної кардіоміопатії (ГКМП)
Генетичні причини ГКМП
У більшості випадків ГКМП спричинена мутаціями генів, що кодують білки серцевого саркомера — структури, відповідальної за скорочення і розслаблення кардіоміоцитів.
Основні гени:
- MYH7 — білок бета-міозинової важкої ланки.
- MYBPC3 — міозин-зв'язуючий білок С.
Рідші гени: TNNT2, TNNI3, TPM1, MYL2, MYL3, ACTC1.
Мутації порушують функцію саркомера, призводячи до гіперконтрактильності, підвищеного споживання енергії, розвитку гіпертрофії, дезорганізації клітин та фіброзу.
Саркомер-негативні випадки
Близько 30–60% пацієнтів мають клінічну ГКМП без виявлених патогенних мутацій. Це вказує на можливу роль ще невідомих генетичних або епігенетичних механізмів.
Варіабельність проявів
Навіть у носіїв однакової мутації хвороба може проявлятися по-різному через вплив інших генів і факторів середовища. Це ускладнює прогнозування перебігу ГКМП.
Спадковість і сімейний анамнез
ГКМП передається аутосомно-домінантно: 50% шанс передати мутацію кожній дитині. У половини пацієнтів виявляється сімейна історія хвороби.
- Оцінка сімейного ризику: важливо зібрати дані про ГКМП, серцеву недостатність, раптову смерть у родичів.
- Скринінг родичів: ЕКГ та ЕхоКГ, починаючи з 10–12 років або раніше при наявності обтяженої історії.
При виявленні мутації можливе каскадне генетичне тестування родичів. Носії мутації потребують регулярного спостереження навіть без симптомів.
Інші фактори ризику розвитку ГКМП
Вік
- Найчастіше маніфестація — у підлітковому або молодому віці.
- ГКМП є провідною причиною раптової смерті у молодих спортсменів.
- Існує форма ГКМП похилого віку (60+ років), яка може мати іншу генетичну основу та перебіг.
Хронічно високий тиск може додатково стимулювати гіпертрофію у генетично схильних пацієнтів та ускладнювати діагностику. Важливо контролювати АТ, уникаючи препаратів, що можуть посилити обструкцію ВТЛШ.
Схожі захворювання ("фенокопії" ГКМП)
Деякі стани імітують клінічну картину ГКМП:
- Серце спортсмена (фізіологічна гіпертрофія).
- Хвороби накопичення: амілоїдоз, хвороба Фабрі, гемохроматоз.
- Інфільтративні хвороби: саркоїдоз.
- Синдромальні стани: синдром Нунан.
- Нервово-м'язові захворювання: атаксія Фрідрейха.
Виключення фенокопій критично важливе для встановлення правильного діагнозу та вибору лікування.
Консультація кардіолога у Львові
Якщо ви хочете дізнатися про стан свого серця - обирайте комплексне обстеження. Воно дозволяє побачити повну картину: від загального стану судин до функції серцевого м’яза.
Вчасне звернення до лікаря про допомогу може врятувати Вам здоров'я, а в більшості випадків - життя.

Якщо ви хочете дізнатися про стан свого серця - обирайте комплексне обстеження. Воно дозволяє побачити повну картину: від загального стану судин до функції серцевого м’яза.
Вчасне звернення до лікаря про допомогу може врятувати Вам здоров'я, а в більшості випадків - життя.
Симптоми та ознаки гіпертрофічної кардіоміопатії (ГКМП)
Гіпертрофічна кардіоміопатія може тривалий час не викликати симптомів. У деяких випадках діагноз встановлюють випадково під час обстеження або скринінгу родичів. Проте у багатьох пацієнтів хвороба проявляється характерними симптомами, що впливають на якість життя.
Найпоширеніші симптоми ГКМП
- Задишка (диспное)
Найчастіший симптом. Спочатку виникає при фізичному навантаженні, з часом може турбувати навіть у спокої. Причина — діастолічна дисфункція та обструкція кровотоку з серця. - Біль у грудях (стенокардія)
Біль стискаючого, пекучого чи ниючого характеру, викликаний ішемією міокарда через недостатнє постачання кисню до потовщеного серцевого м'яза. - Серцебиття
Відчуття перебоїв, прискореного чи сильного серцебиття. Може бути ознакою аритмій: фібриляції передсердь або шлуночкових аритмій.
Інші можливі прояви
- Втома і слабкість
Підвищена втомлюваність навіть при звичних навантаженнях через зниження ефективності роботи серця. - Запаморочення (пресинкопе)
Відчуття легкості в голові або переднепритомний стан, особливо після фізичного навантаження або раптової зміни положення тіла. - Непритомність (синкопе)
Раптова короткочасна втрата свідомості, особливо під час фізичного навантаження, є тривожним симптомом і свідчить про ризик раптової серцевої смерті. - Набряки
Затримка рідини в організмі при розвитку серцевої недостатності: набряки гомілок, стоп, можливий асцит. - Кашель
Сухий кашель, що посилюється у положенні лежачи, може бути ознакою застою крові в легенях.
Коли слід терміново звернутися до лікаря?
Негайно зверніться за медичною допомогою при появі таких симптомів:
- Раптовий сильний біль у грудях.
- Непритомність або сильне запаморочення, особливо під час навантаження.
- Різке посилення задишки або відчуття нестачі повітря.
- Відчуття сильного, нерегулярного серцебиття.
- Будь-які нові або незвичні симптоми, що викликають занепокоєння.
Своєчасне реагування може врятувати життя.
Діагностика гіпертрофічної кардіоміопатії (ГКМП)
Діагностика ГКМП включає збір анамнезу, клінічний огляд і використання інструментальних методів дослідження. Мета — підтвердити діагноз, визначити тип захворювання, оцінити ризики ускладнень.
Клінічний огляд і анамнез
Анамнез: скарги (задишка, біль у грудях, серцебиття, непритомність), індивідуальні хвороби, сімейна історія ГКМП або раптової смерті.
Фізикальний огляд:
- Оцінка набряків, кольору шкіри, набухання вен.
- Пальпація пульсу, верхівкового поштовху.
- Аускультація: грубий систолічний шум вигнання (особливо при обструктивній ГКМП), можливі шуми мітральної регургітації.
- Вимірювання артеріального тиску.
Лабораторні аналізи: загальні аналізи, біохімія, натрійуретичні пептиди.
Основні інструментальні методи
Виявляє ознаки гіпертрофії лівого шлуночка, зміни ST-T, патологічні зубці Q, збільшення лівого передсердя. Важливий метод для первинної діагностики та скринінгу родичів.
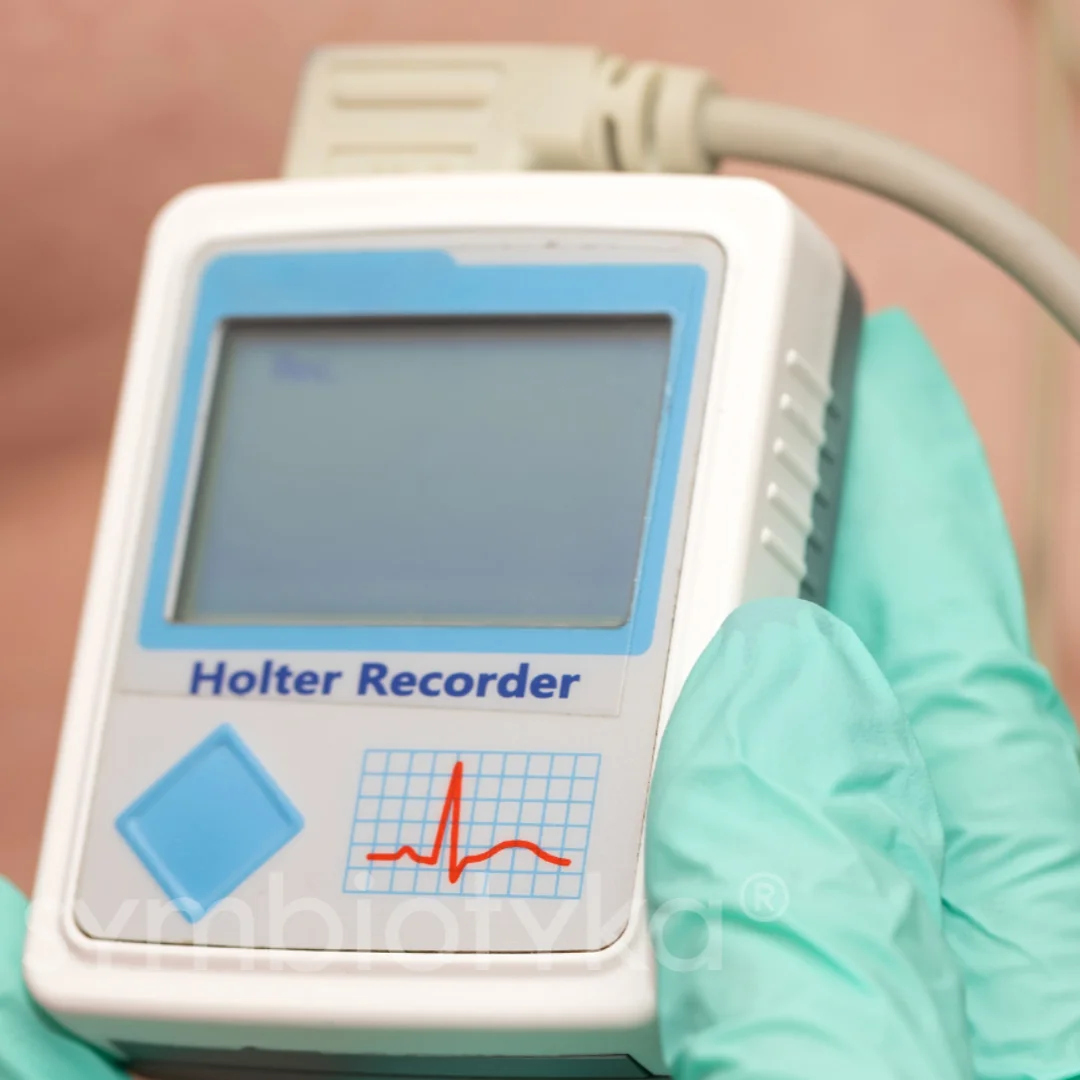
Холтерівське моніторування ЕКГ
Виявляє аритмії:
- Фібриляція передсердь — підвищує ризик інсульту.
- Шлуночкові тахіаритмії — ключовий предиктор раптової серцевої смерті.
Рекомендується при первинній оцінці та періодично для динамічного спостереження.
Підтверджує діагноз: товщина стінки ЛШ ≥15 мм (або ≥13 мм у родичів). Оцінює:
- Морфологію гіпертрофії (асиметрична/симетрична).
- Наявність і ступінь обструкції ВТЛШ.
- Систолічну та діастолічну функцію ЛШ.
- Ступінь мітральної регургітації.
- Розміри лівого передсердя.
ЕхоКГ є золотим стандартом первинної діагностики та моніторингу.
Магнітно-резонансна томографія (МРТ серця)
Використовується для уточнення діагнозу при сумнівних випадках або поганій якості ЕхоКГ. Переваги:
- Деталізація гіпертрофії.
- Виявлення фіброзу міокарда (пізнє накопичення гадолінію, LGE) — важливий предиктор ризику раптової смерті.
- Виявлення апікальної аневризми ЛШ.
МРТ суттєво доповнює стратифікацію ризику у пацієнтів з ГКМП.
Генетичне тестування при ГКМП
Кому рекомендовано: пацієнтам з встановленим діагнозом ГКМП та їх родичам.
Цілі тестування:
- Підтвердити генетичну природу захворювання.
- Провести каскадне тестування родичів для виявлення носіїв мутацій.
Обмеження:
- Негативний результат не виключає діагноз.
- Наявність варіантів невизначеного значення (VUS).
- Виявлення мутації рідко змінює лікування без клінічних проявів.
Генетичне тестування супроводжується консультацією фахівця.
Центр діагностики серця
Ми проводимо діагностику найскладніших серцевих патологій = Ви отримаєте максимально точну картину стану вашої серцево-судинної системи.


.svg)
Сучасні підходи до лікування гіпертрофічної кардіоміопатії (ГКМП)
Лікування ГКМП спрямоване на контроль симптомів, запобігання ускладненням і покращення якості життя. Повністю вилікувати захворювання можливо лише за допомогою трансплантації серця у термінальних стадіях.
Основні цілі лікування
- Полегшення симптомів (задишка, біль у грудях, серцебиття).
- Покращення фізичної активності та якості життя.
- Уповільнення прогресування серцевої недостатності.
- Зниження ризику раптової серцевої смерті (РСС) і тромбоемболій.
План лікування підбирається індивідуально з урахуванням типу ГКМП, симптомів, ризиків та побажань пацієнта.
Медикаментозне лікування ГКМП
Бета-адреноблокатори
- Препарати першої лінії (метопролол, бісопролол, атенолол).
- Знижують частоту серцевих скорочень, покращують діастолічне наповнення, зменшують симптоми та обструкцію.
Блокатори кальцієвих каналів (недигідропіридинові)
- Верапаміл або дилтіазем — альтернатива бета-блокаторам.
- Покращують розслаблення серця.
- Використовуються обережно при обструкції.
Дизопірамід
- Антиаритмічний препарат із негативним інотропним ефектом.
- Зменшує градієнт у ВТЛШ.
- Використовується у комбінації з ББ або БКК.
- Потребує контролю інтервалу QT через ризик аритмій.
Діуретики
- Застосовуються для контролю набряків при серцевій недостатності.
- Використовуються обережно, щоб уникнути посилення обструкції.
Заборонені препарати при обструктивній ГКМП
- Вазодилататори (нітрати, амлодипін, силденафіл).
- Позитивні інотропи (дигоксин, симпатоміметики).
Новітні підходи: інгібітори серцевого міозину
Мавакамтен
- Перший препарат, що цілеспрямовано знижує надмірну скоротливість міокарда.
- Показаний при симптоматичній обструктивній ГКМП (NYHA II–III).
- Покращує симптоми, фізичну витривалість, зменшує градієнт у ВТЛШ.
Моніторинг: регулярне ЕхоКГ для контролю фракції викиду ЛШ. Рекомендовано уникати застосування при ФВ ЛШ <55%.
Інвазивне лікування обструктивної ГКМП
Септальна міектомія
- Хірургічне видалення частини міжшлуночкової перегородки.
- "Золотий стандарт" для важкої обструктивної ГКМП.
- Виконується у спеціалізованих центрах.
Алкогольна септальна абляція (АСА)
- Катетерна процедура, що створює контрольований некроз перегородки за допомогою спирту.
- Альтернатива міектомії для пацієнтів похилого віку або з високим хірургічним ризиком.
Гіпертрофічна кардіоміопатія потребує індивідуального підходу до діагностики, оцінки ризиків і лікування. Медикаментозна терапія залишається основою, новітні препарати — інгібітори міозину — розширили можливості лікування, а септальна редукційна терапія залишається ефективною опцією при тяжкій обструкції.
.png)
.png)

.webp)
Задайте питання в онлайн чаті